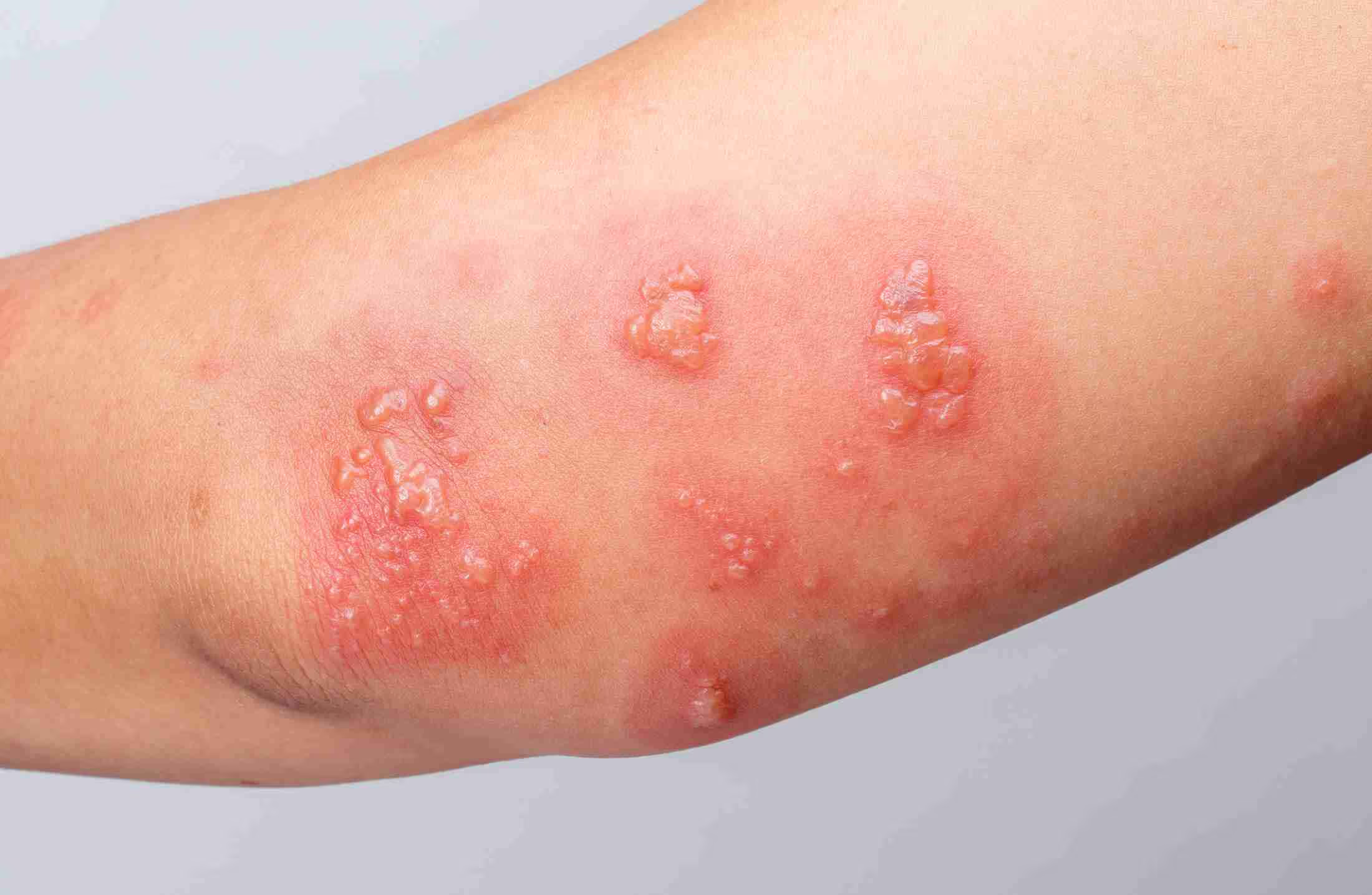
A maioria das doenças raras têm origem genética, enquanto outras são desencadeadas por fatores como infecções, alergias, câncer ou causas ambientais.
O universo das doenças raras é vasto e, por vezes, desconhecido. Isso porque, por serem raras, podem não ser vistas na prática médica ou simplesmente não consideradas durante um diagnóstico. Estima-se que existam mais de 6 mil tipos de doenças raras catalogadas, afetando um percentual significativo da população global, entre 3.5% e 6%.
Esse cenário se traduz em aproximadamente 300 milhões de pessoas em todo o mundo enfrentando condições complexas e desafiadoras. No Brasil, mais de 13 milhões de pessoas são potencialmente afetadas por doenças raras, deixando em evidência uma realidade muitas vezes negligenciada.
Cerca de 80% dessas condições têm origem genética, enquanto outras são desencadeadas por fatores como infecções, alergias, câncer ou causas ambientais. A classificação oficial de "doença rara" é atribuída a condições que afetam um número proporcionalmente pequeno de pessoas em uma população, aproximadamente 1 em 2.000.
Neste artigo, falaremos sobre 15 doenças raras, proporcionando uma visão abrangente dessas condições complexas e muitas vezes esquecidas. Boa leitura!
1. Síndrome Ehlers-Danlos
A Síndrome de Ehlers-Danlos (SED) compreende um conjunto de doenças genéticas raras do tecido conjuntivo, impactando a pele, articulações e vasos sanguíneos. Diversos subtipos da síndrome estão associados a mutações genéticas específicas, sendo a SED causada por alterações nos genes responsáveis pela produção e processamento do colágeno, uma proteína crucial para a estrutura dos tecidos conectivos.
Os sintomas incluem hiperextensibilidade cutânea, articulações hiperflexíveis, fragilidade vascular, dor nas articulações, problemas gastrointestinais e complicações oculares, como miopia e descolamento da retina. Os fatores de risco estão vinculados à herança genética, sendo a SED transmitida de forma autossômica dominante.
O tratamento da síndrome se concentra no gerenciamento dos sintomas individuais, envolvendo fisioterapia, cuidados com a pele, e acompanhamento médico regular para prevenir complicações. A prevalência estatística varia entre os subtipos, mas estima-se que afete, globalmente, aproximadamente 1 em 5 mil a 1 em 20 mil pessoas.
2. Doença de Huntington
A Doença de Huntington é uma condição hereditária e degenerativa do cérebro, resultante de uma mutação no gene HTT localizado no cromossomo 4. A mutação envolve a repetição anormal da sequência de DNA CAG, levando à superprodução da proteína huntingtina mutante. Essa proteína tóxica se acumula nas células cerebrais, especialmente nas regiões associadas ao movimento e cognição.
Os sintomas abrangem distúrbios motores, como movimentos involuntários e coreia, além de declínio cognitivo progressivo, distúrbios psiquiátricos, dificuldades na fala, perda de coordenação e declínio funcional na realização de atividades diárias.
O paciente com Doença de Huntington herdará a mutação do gene HTT de um dos pais, sendo mais comum o início dos sintomas na idade adulta, geralmente entre 30 e 50 anos. Não há cura atualmente para a doença, e o tratamento concentra-se no alívio dos sintomas e na melhoria da qualidade de vida.
Terapias farmacológicas, suporte psicológico e fisioterapia são aplicadas para gerenciar os sintomas. A prevalência da doença varia geograficamente, estimando-se que afete aproximadamente 5 a 10 em cada 100 mil pessoas, especialmente em populações de ascendência europeia, sendo menos comum em populações de ascendência asiática e africana.
3. Fenilcetonúria
A Fenilcetonúria (PKU) é uma doença genética autossômica recessiva causada por mutações no gene PAH, que instrui a produção da enzima fenilalanina hidroxilase essencial. Esta enzima desempenha um papel vital na conversão do aminoácido fenilalanina em tirosina. A ausência ou deficiência dessa enzima leva ao acúmulo tóxico de fenilalanina, prejudicando o desenvolvimento cerebral.
Os sintomas incluem atraso no desenvolvimento cognitivo e motor, convulsões, movimentos involuntários, odor corporal característico devido ao acúmulo de metabólitos de fenilalanina, pigmentação da pele e cabelo, e problemas de comportamento, como hiperatividade e irritabilidade.
A PKU está ligada à genética, exigindo a herança da mutação no gene PAH de ambos os pais para se desenvolver. Pais portadores, mesmo sem sintomas, arriscam ter um filho com a doença. O tratamento inclui uma dieta controlada em fenilalanina, suplementação de tirosina, acompanhamento médico regular, fisioterapia e aconselhamento genético para famílias com histórico de PKU.
A Fenilcetonúria afeta cerca de 1 em 10 mil a 15 mil nascidos vivos. O diagnóstico precoce é vital para evitar danos cerebrais irreversíveis e promover o desenvolvimento adequado do paciente.
.avif)
.jpg)
4. Osteogênese Imperfeita
A Osteogênese Imperfeita (OI), conhecida como "ossos de vidro", é uma condição genética do tecido conjuntivo, afetando predominantemente o colágeno tipo I. A maioria dos casos é resultado de mutações nos genes COL1A1 ou COL1A2, responsáveis pela produção de colágeno. Essas mutações comprometem a qualidade e quantidade do colágeno, resultando em ossos frágeis e suscetíveis a fraturas.
Os sintomas incluem fraturas frequentes com trauma mínimo, esclerótica azul, deformidades ósseas, estatura limitada devido ao crescimento ósseo restrito, problemas dentários e, em casos graves, problemas respiratórios devido à deformidade torácica. Os fatores de risco principais têm origem genética, herdados dos pais, e não há comprovações além desse componente.
O tratamento da OI envolve o controle da dor com analgésicos, cuidados específicos para fraturas, fisioterapia para melhorar a mobilidade e prevenir deformidades, cirurgia em situações que exigem correção de deformidades ou fixação de fraturas, uso de bifosfonatos para fortalecer os ossos e reduzir fraturas, e acompanhamento médico regular para monitorar o crescimento e gerenciar complicações.
Parte do grupo das doenças raras, a prevalência estimada da OI é de 1 em cada 10 mil a 20 mil nascidos vivos, variando em tipos e gravidades da doença.
5. Epidermólise Bolhosa
A Epidermólise Bolhosa (EB), uma doença genética caracterizada pela fragilidade da pele e membranas mucosas devido a mutações em genes que codificam proteínas essenciais para a integridade da pele, como colágeno e queratina. Essas mutações causam falha na ancoragem entre as camadas da pele, resultando na formação de bolhas mesmo com traumas mínimos.
Os sintomas incluem formação recorrente de bolhas na pele e membranas mucosas, cicatrização anormal com deformidades frequentes, bolhas no trato digestivo causando dificuldades alimentares, infecções recorrentes devido à pele danificada e comprometimento do sistema imunológico local, e complicações oculares com bolhas nos olhos podendo resultar em cicatrizes e afetar a visão.
As estratégias de tratamento e prevenção envolvem cuidados específicos para a pele, como manuseio cuidadoso para evitar lesões, uso de curativos especializados, medicamentos para controle da dor associada às bolhas, fisioterapia para manter a mobilidade, prevenção de deformidades e, em casos gastrointestinais, nutrição adequada.
O principal fator de risco é a herança genética, podendo a Epidermólise Bolhosa ser transmitida de forma autossômica dominante ou recessiva, dependendo do subtipo da doença. A prevalência varia conforme o subtipo, estimando-se ocorrer em cerca de 1 em cada 20 mil.
6. Síndrome de Aarskog
A Síndrome de Aarskog é uma condição genética causada por mutações no gene FGD1, localizado no cromossomo X. A síndrome resulta em anomalias faciais distintas, como nariz achatado, sulco nasolabial curto e lábio superior mais largo, baixa estatura, crescimento mais lento, anormalidades genitais como criptorquidia, dedos e mãos curtos e, em alguns casos, deficiência intelectual leve.
A patologia apresenta um risco aumentado em indivíduos com histórico familiar da síndrome, sendo mais comum em homens devido à natureza ligada ao cromossomo X. O tratamento da Síndrome de Aarskog é geralmente sintomático, podendo incluir cirurgia corretiva para anormalidades genitais, intervenções ortopédicas para problemas relacionados e acompanhamento para deficiência intelectual, quando presente.
Não existem medidas específicas para prevenção, já que a síndrome é causada por mutações genéticas. A prevalência da Síndrome de Aarskog varia, estimando-se que afete cerca de 1 em 25 mil a 1 em 80 mil nascidos vivos do sexo masculino, sendo menos incidente em mulheres devido à sua transmissão recessiva ligada ao cromossomo X.
7. Doença de Gaucher
A Doença de Gaucher é uma enfermidade hereditária resultante da deficiência da enzima glucocerebrosidase, levando à acumulação do glucocerebrósido em órgãos. Causada por mutações no gene GBA no cromossomo 1, a deficiência desta enzima causa o acúmulo da substância em células do sistema mononuclear fagocitário.
Os sintomas variam, incluindo hepatoesplenomegalia, anemia, trombocitopenia, osteopenia e osteoporose, com formas raras apresentando comprometimento neurológico. Transmitida de forma autossômica recessiva, exige que ambos os pais carreguem o gene defeituoso para manifestação nos filhos.
Opções de tratamento incluem terapia de reposição enzimática para corrigir a deficiência, terapia de substrato inibidor para reduzir a produção anormal.
Sem meios conhecidos de prevenção, a prevalência varia, sendo mais comum em grupos étnicos específicos, como judeus ashkenazi, com frequência esperada de 1 em 850 nascimentos. Na população geral, a incidência é de 1 em 50 mil até 1 em 100 mil pessoas, com a forma não-neurológica (Tipo 1) sendo mais comum, enquanto as neurológicas (Tipos 2 e 3) são mais raras.
8. Doença de Pompe
A Doença de Pompe, ou glicogenose tipo II, é uma condição genética rara e progressiva resultante da deficiência da enzima alfa-glucosidase ácida, causando acúmulo de glicogênio nos lisossomos celulares, principalmente nos músculos. Mutações no gene GAA, responsável pela produção da enzima, impedem a degradação adequada do glicogênio nos tecidos.
Os sintomas incluem inicialmente fraqueza muscular nos músculos proximais, dificuldades respiratórias devido à fraqueza dos músculos respiratórios, complicações na deglutição devido à fraqueza nos músculos responsáveis pela deglutição, hipotonia em bebês e fadiga pela deterioração muscular progressiva.
Transmitida de forma autossômica recessiva, ambos os pais devem ser portadores do gene defeituoso para a manifestação dos filhos. Tratamentos incluem terapia de reposição enzimática (TRE) para degradação do glicogênio, fisioterapia e reabilitação para função muscular e qualidade de vida, e suporte respiratório em casos avançados.
Não há medidas específicas de prevenção, dada a natureza genética. A prevalência varia em diferentes populações, com incidência estimada de 1 em 40 mil a 1 em 200 mil nascidos vivos, estando, então, dentro do espectro de doenças raras. A forma infantil é mais grave, enquanto a tardia tem progressão mais lenta.
9. Doença de Fabry
A Doença de Fabry, uma condição genética pertencente ao grupo das doenças lisossômicas de depósito, é caracterizada pela deficiência ou ausência da enzima alfa-galactosidase A, levando ao acúmulo progressivo da substância globotriaosilceramida (Gb3 ou GL-3) nas células do corpo e causando danos nos órgãos.
As mutações no gene GLA, que instrui a produção da enzima, são responsáveis pela origem da doença, e sua herança é ligada ao cromossomo X. A doença é hereditária, ligada ao cromossomo X, com homens sendo geralmente mais gravemente afetados, enquanto mulheres portadoras podem apresentar menos sintomas.
Os sintomas da Doença de Fabry incluem dor intensa e queimação nas extremidades, conhecida como dor nos membros, angiokeratomas (lesões avermelhadas ou arroxeadas na pele), problemas gastrointestinais, comprometimento renal com insuficiência progressiva, alterações cardíacas como hipertrofia do ventrículo esquerdo, arritmias e problemas valvulares, além de problemas oculares.
O tratamento envolve a terapia de reposição enzimática (TRE) para administrar a enzima alfa-galactosidase A, reduzindo o acúmulo de Gb3, medicamentos para controlar sintomas, e monitoramento e tratamento de complicações cardíacas e renais. A Doença de Fabry afeta aproximadamente 1 em 40 mil a 60 mil nascidos vivos do sexo masculino.
10. Angioedema hereditário
O Angioedema Hereditário (AEH) é uma doença genética rara e hereditária que provoca episódios recorrentes de inchaço súbito e grave em várias partes do corpo, especialmente nas camadas mais profundas da pele e mucosas. A condição é causada pela deficiência ou disfunção do inibidor de C1 esterase, uma proteína do sistema complemento.
Essa deficiência resulta no aumento da produção de bradicinina, uma substância que induz vasodilatação e aumento da permeabilidade vascular, desencadeando o inchaço característico. O principal fator de risco é a herança genética, geralmente transmitida de forma autossômica dominante, o que significa que uma pessoa afetada tem 50% de chance de transmitir o gene defeituoso a seus filhos.
Os sintomas do AEH incluem inchaço em áreas como rosto, lábios, língua, garganta, mãos, pés, órgãos genitais e trato gastrointestinal, dor abdominal (devido ao inchaço no trato gastrointestinal), desconforto respiratório (se o inchaço envolver a garganta) e coceira e ardência, por vezes acompanhadas de erupções cutâneas.
No que diz respeito ao tratamento do AEH, são utilizados inibidores de bradicinina para controlar os sintomas, bloqueando os efeitos da bradicinina. Além disso, a reposição da proteína deficiente, por meio de concentrados de C1 inibidor, pode ser empregada para prevenir episódios, e agentes antifibrinolíticos podem ser utilizados para reduzir a frequência e a gravidade dos ataques.
Não existem medidas específicas para evitar o desenvolvimento do AEH, uma vez que é uma condição genética. A prevalência exata do Angioedema Hereditário varia em diferentes populações, estimando-se que afete aproximadamente 1 em 50 mil pessoas.
11. Síndrome Wolf-Hirschorn
A Síndrome de Wolf-Hirschhorn é uma doença genética causada por uma deleção no braço curto do cromossomo 4, mais especificamente na região 4p16.3. Essa síndrome impacta significativamente o desenvolvimento do corpo e do cérebro, resultando em características físicas e cognitivas distintas.
Os sintomas e características da Síndrome de Wolf-Hirschhorn podem variar, mas geralmente incluem retardo no desenvolvimento motor e cognitivo, deficiência intelectual que varia de leve a grave, características faciais distintas como testa estreita, olhos amplamente espaçados, nariz pequeno e lábio superior curto, baixa estatura, possíveis anomalias cardíacas, convulsões em alguns casos e problemas renais, como anomalias nos rins.
O principal fator de risco está associado à causa genética, ocorrendo muitas vezes de forma esporádica, embora casos de herança dos pais também sejam observados. Devido à sua causa genética, não existe uma cura para a síndrome. O tratamento é sintomático e abrange diversas especialidades médicas, além de outras áreas da saúde, como fisioterapia, terapia ocupacional e suporte educacional.
O objetivo do tratamento é melhorar a qualidade de vida e o funcionamento diário do indivíduo afetado. Estima-se que a Síndrome de Wolf-Hirschhorn ocorra em aproximadamente 1 em 50 mil nascidos vivos. A incidência real pode ser subestimada devido à variabilidade dos sintomas e ao desconhecimento de alguns casos.
12. Doença de Crohn
A Doença de Crohn, uma forma de doença inflamatória intestinal (DII), é uma enfermidade inflamatória crônica que pode afetar qualquer parte do trato gastrointestinal. A causa exata envolve complexa interação entre fatores genéticos, ambientais e imunológicos, destacando predisposição genética e resposta imunológica anormal às bactérias intestinais.
Os sintomas são bastante variados, e podem incluir diarreia persistente, dor abdominal, perda de peso, fadiga, febre, fissuras anais e úlceras bucais. Os fatores de risco incluem histórico familiar da doença, idade (geralmente diagnosticada entre 15 e 35 anos), tabagismo (que aumenta o risco e pode agravar a doença) e histórico de infecções gastrointestinais.
As opções de tratamento e gestão da Doença de Crohn envolvem medicamentos anti-inflamatórios para controlar a inflamação, imunossupressores para reduzir a resposta imunológica, terapia biológica que utiliza anticorpos para modular a resposta imunológica, e cirurgia em casos de complicações, como obstruções intestinais ou perfurações.
A prevalência da doença é mais significativa em países desenvolvidos, especialmente na Europa e América do Norte, estimando-se que afete cerca de 1 a 3 em cada 100 mil pessoas – dentro do espectro das doenças raras.
13. Esclerose Múltipla (EM)
A esclerose múltipla (EM) é uma doença autoimune que afeta o sistema nervoso central, resultando no ataque do sistema imunológico à mielina, a camada protetora dos nervos. Embora as causas exatas permaneçam desconhecidas, fatores genéticos e ambientais parecem estar relacionados na suscetibilidade à doença.
Os sintomas da EM são amplos, e abrangem fadiga, dificuldades de coordenação, visão turva, dormência ou formigamento, problemas de equilíbrio, fraqueza muscular e, em casos mais graves, incapacidade. Fatores de risco identificados incluem histórico familiar da doença, idade (com diagnósticos frequentes entre 20 e 40 anos), maior prevalência em mulheres e influências ambientais, como exposição à luz solar e deficiência de vitamina D.
Atualmente, não há cura para a esclerose múltipla, mas diversas abordagens terapêuticas visam reduzir os sintomas, controlar exacerbações e atrasar a progressão da doença. Medicamentos imunomoduladores são comumente prescritos, com terapias de suporte, incluindo fisioterapia e medicamentos para gerenciar sintomas específicos.
A prevalência global da esclerose múltipla pode variar de 2 a 150 casos por 100 mil habitantes; estima-se que cerca de 2,8 milhões de pessoas em todo o mundo tenham EM, sendo mais comum em regiões afastadas do equador e predominantemente afetando pessoas de ascendência europeia.
14. Síndrome de Hermansky-Pudlak
A Síndrome de Hermansky-Pudlak (HPS) é uma condição genética que afeta vários órgãos e sistemas do corpo, caracterizada por uma combinação de albinismo, distúrbios de coagulação e disfunções em organelas celulares conhecidas como lisossomos e melanosomas.
As causas da síndrome residem em mutações em vários genes, dependendo do tipo específico da HPS, afetando a função dessas organelas e resultando em distúrbios em diferentes sistemas corporais.
Os sintomas incluem albinismo, problemas de sangramento devido a distúrbios do sangue plaquetários e deficiência de fator de coagulação, complicações pulmonares como fibrose em casos específicos, problemas gastrointestinais, complicações renais e problemas oculares, como estrabismo e diminuição da acuidade visual.
A síndrome é transmitida de forma autossômica recessiva, exigindo que ambos os pais sejam portadores do gene defeituoso para que a condição se manifeste em seus filhos. As opções de tratamento incluem transfusões sanguíneas para problemas de coagulação, suporte respiratório em casos de fibrose pulmonar grave, e cirurgia, quando necessário, para corrigir complicações como colite.
A prevalência da Síndrome de Hermansky-Pudlak varia entre as populações, sendo mais comum em comunidades com maior consanguinidade. Estima-se uma prevalência geral de aproximadamente 1 em 500 mil a 1 em 1 milhão de nascidos vivos.
15. Progeria
A Progeria, também conhecida como Síndrome de Hutchinson-Gilford, é uma rara doença genética caracterizada pelo envelhecimento acelerado em crianças. A condição é originada por uma mutação espontânea no gene LMNA, resultando na produção de uma forma anormal da proteína lamin A.
Essa proteína compromete a integridade estrutural das células, desencadeando um processo de envelhecimento celular prematuro. As crianças com Progeria apresentam sinais de envelhecimento rápido, incluindo rugas na pele, perda de cabelo, articulações rígidas e outros sintomas associados a idades avançadas.
A principal causa da Progeria é genética, resultante de mutações espontâneas no gene LMNA, e não é uma condição herdada dos pais. Os sintomas incluem não apenas o envelhecimento acelerado, mas também problemas cardiovasculares, baixo crescimento e peso, rigidez nas articulações e perda de gordura subcutânea.
Não há fatores de risco específicos associados à condição, uma vez que sua origem está na mutação genética. Atualmente, não há uma cura conhecida para a Progeria, e o tratamento visa aliviar os sintomas e melhorar a qualidade de vida dos pacientes. Terapias de suporte, como o tratamento de problemas cardíacos, fisioterapia e medicamentos sintomáticos, são aplicadas.
A prevalência da Progeria é extremamente baixa, estimada em 1 em 20 milhões de nascimentos, afetando indivíduos de todas as raças, etnias e sexo.
Invista na sua atualização profissional com a Afya Educação Médica!
A trajetória das pessoas que enfrentam doenças raras é multifacetada, desde os desafios do diagnóstico até a busca por tratamentos adequados. Enquanto alguns distúrbios raros se manifestam desde o nascimento, outros surgem apenas na fase adulta.
Diante deste amplo cenário, a procura por identificação e tratamento se configura frequentemente como uma jornada árdua, necessitando de uma abordagem interdisciplinar e de considerável paciência. Muitos pacientes recorrem a diversos profissionais de saúde antes de obterem uma compreensão precisa de sua condição, evidenciando a complexidade e a demora associadas ao processo.
Vale ressaltar que a identificação precoce e a intervenção multidisciplinar continuam a ser elementos cruciais para a otimização do desenvolvimento e do bem-estar dos indivíduos afetados pelas doenças raras.
Assine a nossa newsletter e receba posts exclusivos e atualizações sobre Medicina diretamente no seu e-mail!




.png)

.png)
.png)
